1) Vieillissement : mécanismes fondamentaux, interactions avec l’environnement et recherche de composés anti-âge
Le vieillissement est un processus qui reste encore mal compris en dépit d’importants progrès réalisés ces trois dernières décennies. De plus, ses conséquences délétères, avec en particulier l’incidence croissante des pathologies liées à l’âge, pèsent de plus et plus au niveau socio-économique compte-tenu des évolutions démographiques des sociétés humaines. Il est donc critique de progresser dans notre compréhension des mécanismes fondamentaux impliqués dans le vieillissement et ses pathologies associées.
Nous développons pour cela des approches de génétique et de génomique fonctionnelle chez la drosophile. Nous avons ainsi mis en évidence des réseaux géniques impliqués dans le vieillissement cardiaque. Nous nous intéressons également à l’impact de facteurs environnementaux modulant le métabolisme et la longévité. Nous avons récemment observé des interventions environnementales conférant des augmentations de longévité se répercutant sur plusieurs générations et cherchons à présent à identifier les mécanismes impliqués dans cet effet mémoire.
En parallèle, une collaboration avec la société chinoise Infinitus nous a permis d’identifier des composés de la médecine traditionnelle chinoise présentant la capacité de contrecarrer des processus liés au vieillissement (résistance aux stress, dysfonction cardiaque, réduction de l’activité locomotrice…) et ayant un effet protecteur en contexte de pathologie neurodégénérative.
2) Pathologies neurodégénératives et cardiaques
Nos projets exploitent la puissance du modèle Drosophile pour modéliser des pathologies dégénératives, en explorer les mécanismes physiopathologiques et tester des stratégies thérapeutiques, en s’appuyant sur les capacités de criblages génétiques et pharmacologiques permises chez cet organisme.
2-a. Maladies à expansions de triplets de polyglutamine (maladie de Huntington (MH) et Ataxie spino-cérébelleuse de type 3 (SCA3))
Ces deux maladies sont liées à des amplifications du triplet CAG, codant pour la glutamine, dans les régions codantes des gènes de la Huntingtine et de l’ataxine 3 respectivement. Elles sont toutes deux des protéinopathies, caractérisées par l’apparition d’agrégats intranucléaires et la dégénérescence de régions spécifiques du système nerveux central comme le cervelet, le pons, le cortex ,…
Dans le cadre du programme européen TreatPolyQ, nous avons collaboré avec les équipes de L. Pereira de Almeida (Coimbra, Portugal) et T. Schmidt (Tübingen, Allemagne) pour étudier le rôle de certains microARNs et de facteurs d’exportation nucléaire dans la pathologie SCA3. Grâce au modèle Drosophile, nous avons ainsi identifié un transporteur nucléaire de la famille des karyophérines, KPNA3 (aussi appelé Importin subunit alpha-4) comme étant un élément clé dans la localisation nucléaire de l’ataxine 3. Ainsi, la diminution de l’expression de KPNA3 relocalise la protéine SCA3 mutante dans le cytoplasme des cellules et améliore la dégénérescence observée dans l’œil de la drosophile en contexte pathologique, faisant de KPNA3 une potentielle cible thérapeutique d’intérêt.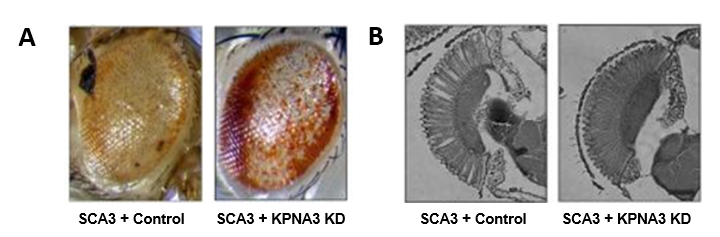 Figure 1 : A. En conditions contrôles (SCA3 + Control), l’expression de l’ataxine 3 pathologique dans l’œil de drosophile entraine des défauts morphologiques : dépigmentation, nécrose, désorganisation générale (image de gauche), que la diminution d’expression de KPNA3 (SCA3 + KPNA3 KD) permet d’améliorer (image de droite) : œil partiellement repigmenté et en partie réorganisé. B. L’observation de la rétine de ces mouches montre une nette amélioration de la morphologie des photorécepteurs lors de la diminution de l’expression de KPNA3 (image de droite) comparé aux conditions contrôles, où l’on constate des vacuoles dans la rétine, ainsi qu’une désorganisation des photorécepteurs.
Figure 1 : A. En conditions contrôles (SCA3 + Control), l’expression de l’ataxine 3 pathologique dans l’œil de drosophile entraine des défauts morphologiques : dépigmentation, nécrose, désorganisation générale (image de gauche), que la diminution d’expression de KPNA3 (SCA3 + KPNA3 KD) permet d’améliorer (image de droite) : œil partiellement repigmenté et en partie réorganisé. B. L’observation de la rétine de ces mouches montre une nette amélioration de la morphologie des photorécepteurs lors de la diminution de l’expression de KPNA3 (image de droite) comparé aux conditions contrôles, où l’on constate des vacuoles dans la rétine, ainsi qu’une désorganisation des photorécepteurs.
Nous avons également développé un modèle cardiaque de la maladie de Huntington, et plus récemment un modèle glial inductible. Ce dernier complète les modèles neuronaux existants et nous permet d’explorer les conséquences respectives de l’expression de la Huntingtine pathologique dans les différents types cellulaires du système nerveux central (glie ou neurones) sur différents paramètres physiologiques (longévité, activité locomotrice, rythme circadien ..). Nous avons ainsi identifié de profondes différences entre les modèles neuronaux et gliaux de la MH, ce qui illustre la complexité de la maladie. Nous avons réalisé un crible génétique afin d’identifier les voies de signalisation impliquées et, sur ces bases, explorer de nouvelles stratégies thérapeutiques. Nous avons ainsi mis en évidence que la déplétion partielle du facteur de transcription nej/dCBP dans les cellules gliales était protectrice et permettait de restaurer l’activité locomotrice des mouches MH et d’augmenter leur durée de vie. Ainsi, dCBP contribue au développement de la pathologie dans les cellules gliales, contrairement à ce qui avait été observé dans les neurones. Nos données suggèrent donc que des approches combinatoires associées à un ciblage spécifique des tissus pourraient être nécessaires pour découvrir des thérapies efficaces contre la MH.
De nouveaux modèles Drosophile de MH, développés grâce aux techniques CRISPR/Cas9, sont également actuellement en cours d’étude.
2-b. Ataxie de Freidreich (AF)
L’AF est une pathologie mitochondriale qui touche principalement le système nerveux et le cœur. Elle est due à une expansion de triplets GAA dans le premier intron du gène FXN entrainant la diminution de son expression. FXN code la frataxine, une petite protéine mitochondriale impliquée dans la synthèse des centres fer-soufre et très conservée au cours de l’évolution. Nous utilisons la Drosophile pour mieux comprendre les mécanismes impliqués dans cette pathologie et rechercher de nouvelles pistes thérapeutiques.
Nous avons tout d’abord établi un modèle cardiaque de l’AF chez la Drosophile et montré que l’inactivation coeur-spécifique de la frataxine conduit à une dilatation cardiaque avec perte de contractilité des cardiomyocytes et altération des sarcomères. Nous avons réalisé des cribles pharmacologiques sur ce modèle cardiaque et identifié plusieurs molécules à effet cardioprotecteur dont le bleu de méthylène.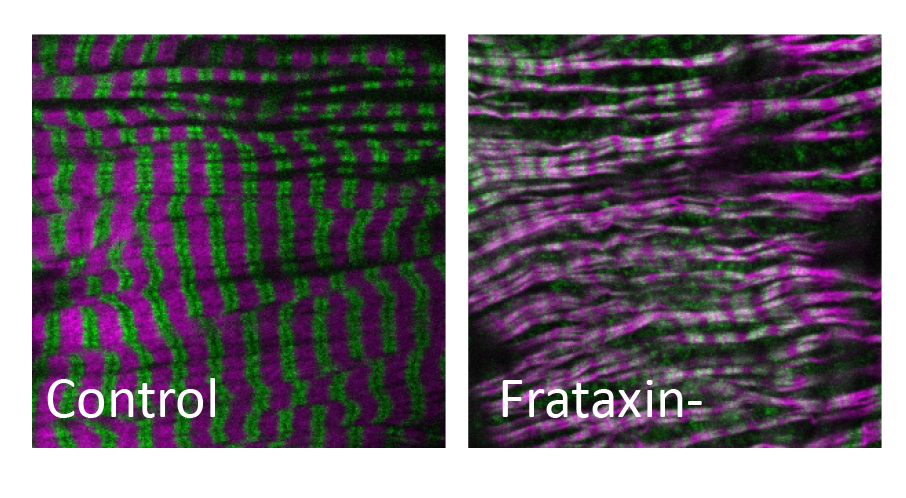 Figure 2 : Altération des sarcomères (Actine en magenta et Myosine en vert) dans des cardiomyocytes de mouches dans lesquels la frataxine a été inactivée.
Figure 2 : Altération des sarcomères (Actine en magenta et Myosine en vert) dans des cardiomyocytes de mouches dans lesquels la frataxine a été inactivée.
Nos approches actuelles s’appuient sur de nouveaux modèles de l’AF chez la Drosophile obtenus par humanisation du gène de la frataxine de Drosophile. Pour cela, nous avons inséré dans l’intron du gène Drosophile de la frataxine, des séquences introniques du gène humain portant des expansions GAA de tailles variées. Ces expansions entrainent une diminution de l’expression du gène et reproduisent des symptômes associés à la pathologie humaine (déficits locomoteurs et cardiaques, viabilité réduite). Nous exploitons actuellement ces nouveaux modèles en développant des approches pharmacologiques (criblages secondaires de chimiothèques et recherche de peptides thérapeutiques), en nous appuyant sur des collaborations nationales et internationales et grâce au soutien de l’association française AFAF (Association française de l’Ataxie de Friedreich) et de l’association américaine FARA (Friedreich Ataxia Research Alliance).
2-c. Maladie de Parkinson
La maladie de Parkinson (MP) est la deuxième maladie neurodégénérative la plus fréquente après la maladie d’Alzheimer. On estime ainsi qu’elle touche 1% des plus de 65 ans. Elle se caractérise par la perte lente et progressive des neurones à dopamine situés dans la substance noire du cerveau, ce qui entraine des problèmes de contrôle des mouvements mais aussi des troubles cognitifs. Il n’existe actuellement aucun traitement curatif pour cette pathologie, ce qui en fait un véritable enjeu de santé publique. Dans ce contexte, nous utilisons des modèles drosophile de la MP afin d’identifier des mécanismes impliqués dans la pathogenèse de cette maladie et qui peuvent être utilisés comme cibles thérapeutiques. Récemment, le gène LRRK2, qui rend compte tant de formes sporadiques que familiales de la MP, a été identifié comme une cible thérapeutique clé. Plus spécifiquement, l’état de phosphorylation de LRRK2 semble influencer son fonctionnement physiologique et pathologique. Nous avons donc développé des collaborations nationales afin de déterminer et de valider des phosphorégulateurs de LRRK2 permettant de moduler son état de phosphorylation et qui pourraient constituer de potentiels candidats médicaments.
Ces travaux collaboratifs ont été initiés dans le cadre d’un financement Emergence en Recherche Idex Université de Paris et bénéficient du soutien de l’ANR (projet PARK-PEP).
2-d. Criblage de variants identifiés chez des patients atteints de cardiomyopathies.
La Drosophile s’est imposée ces dernières années comme un modèle pertinent pour identifier des gènes impliqués dans le développement de cardiomyopathies et étudier les mécanismes impliqués dans leur pathogénicité. Grâce à un système d’imagerie cardiaque in vivo développé dans l’équipe, nous utilisons cet organisme pour étudier des gènes pour lesquels des variants ont été identifiés chez des patients par des approches génomiques réalisées par nos collaborateurs. Cette approche permet un criblage fonctionnel in vivo de gènes candidats afin d’apporter rapidement des arguments en faveur ou non de leur pathogénicité. Figure 3 : Imagerie cardiaque in vivo chez la Drosophile.
Figure 3 : Imagerie cardiaque in vivo chez la Drosophile.
Des Drosophiles anesthésiées exprimant une protéine fluorescente GFP dans le coeur sont placées sur une lame sous stéréomicroscope. La lumière de fluorescence émise est captée à travers la cuticule dorsale par une caméra à haute vitesse, ce qui permet d’extraire les paramètres cardiaques.
3) Recherche de biomarqueurs d’efficacité pour le suivi de patients atteints de maladies neurodégénératives : application en cas de traitements
Des dérégulations du métabolisme des monocarbonés sont associées à une incidence accrue de plusieurs maladies chroniques, comme l’hyperhomocystéinémie, la maladie d’Alzheimer et d’autres maladies métaboliques comme le diabète de type 2 et l’obésité. Bien que les mécanismes moléculaires qui déclenchent la maladie d’Alzheimer ne sont pas entièrement compris, de récentes données suggèrent que la dyslipidémie peut contribuer à sa progression. Certains patients avec trisomie 21 développent à l’âge de 30-40 ans une démence comparable à celle de la maladie d’Alzheimer. Le vieillissement pathologique chez le sujet avec trisomie 21 est associé au syndrome démentiel qui combine à des degrés variés des troubles des fonctions cognitives et du comportement modifiant la personnalité. Ces patients présentent également une dyslipidémie avec des dérégulations du métabolisme des monocarbonés. Parmi les gènes localisés sur le chromosome 21, l’expression de DYRK1A, une sérine thréonine kinase, et de la CBS (cystathionine beta synthase), enzyme impliquée dans le métabolisme des monocarbonés, est dérégulée dans la maladie d’Alzheimer. Nous avons démontré une relation entre DYRK1A et la CBS et leur implication dans le métabolisme des monocarbonés, du cholestérol et de l’insuline, métabolismes dérégulés dans la maladie d’Alzheimer, l’hyperhomocystéinémie, la trisomie 21 et le diabète de type 2. Cibler ces deux protéines pour un traitement pharmacologique nécessite une meilleure compréhension de leur fonction et de leurs partenaires en interaction. La recherche d’interactants, en collaboration avec le Dr JM Camadro (Institut Jacques Monod, UMR CNRS 7592), a permis l’identification de protéines déjà montrées comme potentiels biomarqueurs dans le sérum de patients atteints de la maladie d’Alzheimer. Ces différents biomarqueurs sont actuellement analysés dans le plasma et le liquide céphalo-rachidien de patients atteint de maladie d’Alzheimer et de patients porteurs de trisomie 21 avec ou sans démence et au stade prodromal (en collaboration avec le Dr Anne-Sophie Rebillat (Institut Jérôme Lejeune, Paris) et le Pr J Fortea (Memory Unit, Department of Neurology, Hospital de la Santa Creu i Sant Pau-Biomedical Research Institute Sant Pau-Universitat Autònoma de Barcelona, Barcelona, Spain). L’évaluation de ces biomarqueurs dans le sérum des patients combinée à des paramètres d’imagerie et des marqueurs du liquide céphalo-rachidien devra permettre de développer un test à visée pronostique/diagnostique dans le but d’identifier les personnes à haut risque de développer la maladie d’Alzheimer et qui pourront bénéficier de traitements.
Outre la validation de nouveaux biomarqueurs sanguins, l’évaluation cerveau-plasma dans des modèles de souris et de rats nous permet d’étudier les mécanismes neurobiologiques sous-jacents à la dérégulation de ces biomarqueurs. Dans ce but, nous cherchons à démontrer leur rôle dans la progression de la maladie d’Alzheimer par l’utilisation du premier modèle de rat inductible et progressif de la maladie, en collaboration avec le Dr Jérôme Braudeau (AgenT).
Ces modèles murins nous permettent également de rechercher des traitements ciblant DYRK1A et la CBS par approche computationnelle et tests in vitro et in vivo (dans le cadre d’un consortium, Fig 3).
 Figure 4: Consortium pour la recherche d’inhibiteurs par approche computationnelle et tests in vitro et in vivo
Figure 4: Consortium pour la recherche d’inhibiteurs par approche computationnelle et tests in vitro et in vivo
Dans le cadre de la recherche de traitements, ces biomarqueurs sont également utilisés au stade préclinique afin d’analyser les effets de traitements à un stade précoce, que nous développons actuellement selon deux grandes voies :
. une voie ciblant plus spécifiquement l’activité de DYRK1A avec un inhibiteur pharmacologique spécifique (en collaboration avec le Dr Yann Hérault (Institut de Biologie Moléculaire et Cellulaire, UMR 7104 du CNRS, U1258 INSERM, Strasbourg) et le Dr Laurent Meijer (Perha Pharmaceuticals, Roscoff)).
.une voie proposant de tester une molécule au stade embryonnaire (en collaboration avec le Pr François Vialard (Université Paris-Saclay, UVSQ, INRAE, ENVA, BREED, 78350, Jouy-en-Josas, France) et le Pr Anne-Claude Camproux (Université de Paris, BFA, UMR 8251, CNRS, ERL U1133, Inserm)).
Dans le cadre d’une étude clinique, un premier essai de phase II, randomisée avec placebo, qui a été conduit par différentes équipes dont notre groupe, a permis de mettre en évidence que l’EGCG améliore les capacités cognitives et d’apprentissage de jeunes adultes porteurs de trisomie 21 (Fig. 4). Cette étude est actuellement poursuivie chez de jeunes enfants porteurs de trisomie 21, après avoir déterminé la dose et l’efficacité à l’aide d’un modèle préclinique, dans le cadre d’une étude de phase II en collaboration avec le Dr Cécile Cieuta-Walti (Institut Jérôme Lejeune, Paris) et le Pr Rafael de la Torre (IMIM-Hospital del Mar Medical Research Institute, Barcelona).
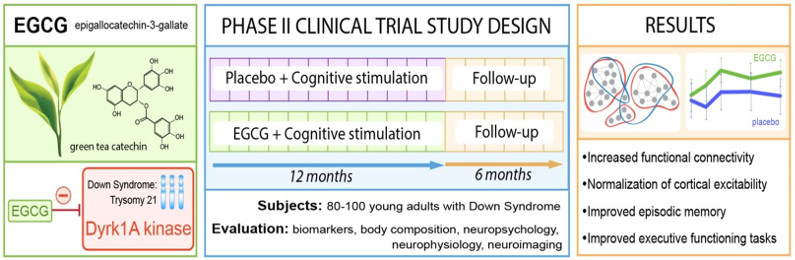
Figure 5 : Principe de l’essai clinique de phase 2 avec l’EGCG, un inhibiteur de DYRK1A, chez des patients porteurs de trisomie 21

