Cinq grandes thématiques sont abordées au laboratoire :
1- Mécanismes physiopathologiques associés aux filaments intermédiaires cytosoliques ou nucléaires et leurs protéines partenaires dans les myopathies:
A/ Myopathies liées à des mutants de protéines nucléaires (Responsable: B. Buendia): Nathalie VADROT et Dr Brigitte BUENDIA.
Des mutations dans le gène codant pour les lamines de type A, protéines nucléaires de la famille des filaments intermédiaires, sont responsables de diverses pathologies. Nous en étudions les mécanismes moléculaires sous-jacents, en collaboration avec des cliniciens (équipe du Pr Corinne Vigouroux ; Paris) ainsi qu’avec des experts i) de la structure 3D de complexes de protéines nucléaires (équipe du Dr Sophie Zinn-Justin ; Saclay) et ii) de domaines de la chromatine associés aux lamines, nommés LADs (équipe du Pr Philippe Collas ; Oslo-Norvège). Le développement de modèles cellulaires et animaux nous a permis de révéler que des lamines de type A mutées en certains sites responsables de myopathie avaient une capacité différente de celle des lamines sauvages pour s’assembler, lier l’ADN et/ou des protéines partenaires, avec des répercussions sur la morphologie du noyau (Figure 1), l’organisation / l’expression du génome, associés à des défauts de prolifération/différenciation des cellules ou de typage des fibres musculaires. Notre projet actuel porte sur une protéine partenaire des lamines de type A, LAP2, dont nos collègues (équipe du Dr Pascale Richard ; Paris) viennent d’identifier des mutants associés à des pathologies du muscle cardiaque.
Publications de l’année : Paulsen et al. 2017 ; Barateau et al. 2017
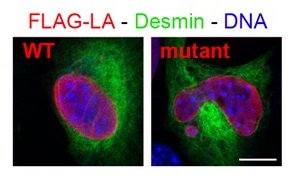
Figure 1: Immunofluorescence de myoblastes exprimant la lamine A (LA) étiquetée FLAG soit sauvage (WT) ou mutée (Mutant) (responsable de dystrophie musculaire congénitale) ; from Barateau et al. 2017.
B/ Desmin-related myopathies (desminopathies) (Responsable: S. Batonnet-Pichon) : Dr Sabrina BATONNET-PICHON, Dr Alain LILIENBAUM, Dr Eva CABET, Florence DELORT, Coralie HAKIBILEN.
Les desminopathies sont des pathologies rares touchant le système neuromusculaire. Elles sont transmises généralement selon un mode autosomal dominant et apparaissent le plus souvent à l’âge adulte. Elles se caractérisent particulièrement par l’accumulation d’agrégats de desmine, un filament intermédiaire localisé dans le sarcoplasme des cellules musculaires squelettiques et cardiaques. A ce jour plus de soixante-dix mutations associées à cette pathologie ont été décrites sur le gène de la desmine. Notre laboratoire a historiquement participé à l’identification d’au moins une vingtaine d’entre-elles. Actuellement, nous étudions plus particulièrement les mutations faux-sens D399Y, E413K, R406W (Figure 2). Nos approches sont très orientés multidisciplinaires, ce qui nous permet d’enrichir nos perspectives d’études de la desmine.
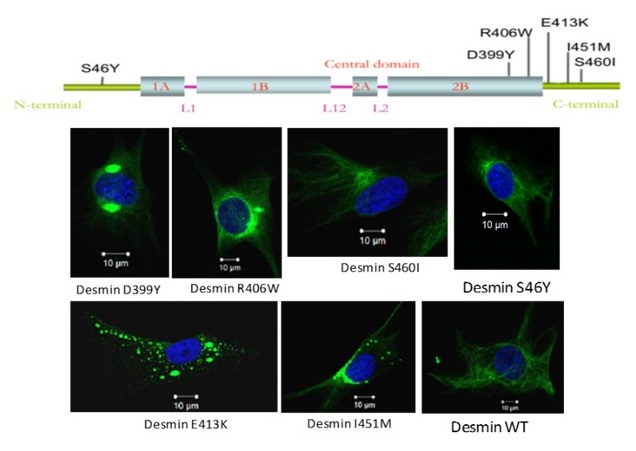
Figure 2: Représentation schématique de la desmine et de la localisation des principales mutations étudiées au laboratoire.
Ainsi pour étudier les mécanismes moléculaires qui leur sont liés, nous avons développé différents modèles qui vont de clones stables dérivés de myoblastes C2C12, exprimant la desmine sauvage ou mutée de manière inductible, jusqu’aux modèles animaux par injection d’AAV (Comité d’éthique #5441 CEF) ou plus récemment de Souris knock-in R405W (homologue murine de la mutation R406W humaine, Comité d’éthique CEB-16-2016). A l’aide de ces modèles, nous explorons donc différents mécanismes moléculaires liées à ces pathologies tels que :
a- Premièrement, nous avons choisi d’étudier un modèle murin de myopathie myofibrillaire pour mieux comprendre les étapes précoces de la mise en place de la maladie, et afin de pouvoir évaluer des stratégies thérapeutiques. La mutation R406W de la desmine humaine produit des effets sévères et précoces chez les patients, en particulier sur les muscles squelettiques des membres, cardiaques et respiratoires. La souris knock-in permet le remplacement du gène endogène par le gène muté et constitue l’approche la plus physiologique. Les souris hétéro- et homozygotes présentent une accumulation anormale de desmine à la membrane sarcoplasmique des muscles striés, en particulier le Tibialis Anterior et le Soléaire. Cette accumulation s’aggrave avec l’âge. De plus, les souris homozygotes sont plus petites et meurent à l’âge de 3 mois environ (Figure 3). Des études en cours vont permettre de préciser si cette létalité est due à des problèmes cardiaques, respiratoires, ou sont la conséquence de défauts critiques d’autres organes.
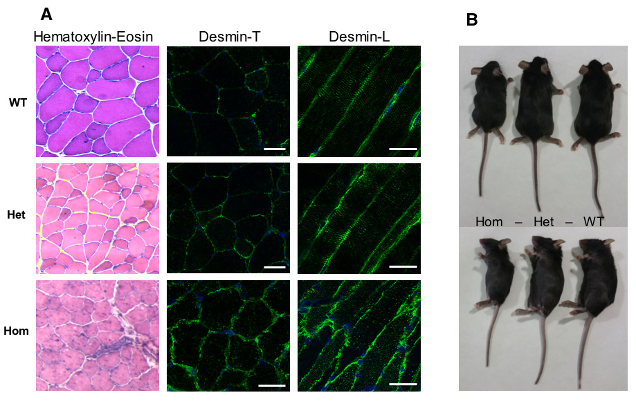
Figure 3: Présentation du modèle murin Knock-in desR405W (homologue de la mutation humaine R406W) : les souris homozygotes présente une altération structurelle des fibres musculaire ainsi qu’une diminution drastique de taille.
b- Deuxièmement, nous étudions les propriétés mécaniques des cellules exprimant la desmine sauvage ou mutée grâce à deux fortes collaborations établies avec des biophysiciens dans le cadre d’une ANR en cours :
- F. Briki (LPS, Orsay) : nous avons pu montrer que l’expression du mutant D399Y de la desmine présente une altération de la réorientation cellulaire sous contrainte (Leccia et al, 2013). Nous avons également étudié la rigidité des cellules exprimant la desmine D399Y à l’aide de la microscopie à force atomique (Article en révision)
- S.Hénon (MSC, Paris Diderot) : nous avons ainsi récemment montré que la surexpression de la desmine sauvage par électroporation dans les C2C12 est capable de rigidifier de manière globale les cellules. Au contraire, la mutation E413K ne provoque pas cette rigidification supposant une perte de fonction. De plus, à l’aide du Rhéomètre à cellule unique (A. Asnacios, MSC, Paris Diderot), et de microscopie à traction de Force (B.Kalman, Grenoble), nous avons pu déterminer que la surexpression de la desmine E413K dans les C2C12 diminue leur capacité à générer une force contractile (charrier el al, 2016). Cet effet est associé à une altération du nombre et de la longueur des fibres de stress d’actine (Charrier et al, 2016). Enfin, nous avons également montré que la desmine joue un rôle au niveau des propriétés rhéologiques cytoplasmiques des cellules et non pas corticales, à l’aide de pinces optiques et magnétiques (Charrier et al, soumis).
c- Plus récemment avec l’arrivée de C.Hakibilen (Doctorante de BioSPC), toujours en collaboration avec S.Hénon, (MSC Paris Diderot), nous avons commencé un nouveau projet (financement AFM) sur le rôle de la desmine et l’impact des mutations sur l’adhésion cellule-matrice (figure 4, gauche), et sur l’adhésion cellule-cellule (collaboration en cours de mise en place avec RM Mège, IJM, Paris Diderot). Ce projet repose en partie sur l’utilisation de l’imagerie TIRFM (Total internal reflection fluorescence microscopy, BFA), nous permettant de quantifier plus spécifiquement les adhésions focales (leur nombre, leur aire…). Ces analyses seront réalisée non seulement sur des C2C12 (figure 4, droite), mais également de cellules satellites primaires issues des souris desKO et desKI du laboratoire
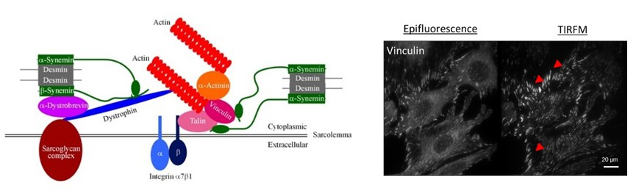
Figure 4 : A gauche, représentation schématique de la desmine dans une adhésion focale de cellule cardiaque. A droite, visualisation des adhésions focales de cellules C2C12 sur fibronectine par imagerie de type épifluorescence ou TIRFM (BFA, plateau technique).
d- D’un point de vue biochimique, nous étudions les interactions avec différents partenaires tels que les protéines Dux 4 et Dux4C en collaboration avec F.Coppée (Mons, Belgique).
e- Nous avons également pu mettre en évidence que le stress oxydatif, ou thermique induit l’apparition de l’agrégation du mutant de desmine D399Y in cellulo (Segard et al, 2013, Figure 5). Cet effet a pu être confirmé pour d’autres mutants tels que Q389P, ou R406W mais n’est pas généralisable à l’ensemble des mutations de la desmine, ni de la région 2B (Delort F. et al, Article en rédaction).
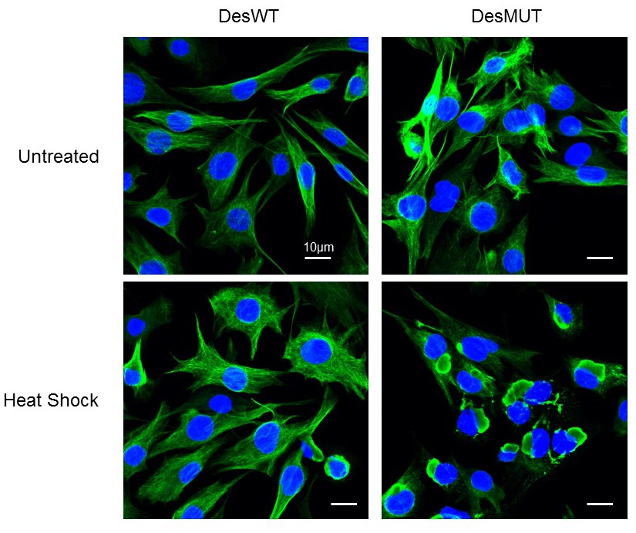
Figure 5 : Effet d’un traitement Heat-shock sur l’agrégation de la desmine D399Y dans les clones stables inductibles. (Image réalisé avec le microscope confocal LSM700 de la plateforme d’Imagerie BFA)
f- Enfin, sachant qu’à l’heure actuelle aucun traitement n’est disponible pour les patients atteints de desminopathies, nous recherchons activement des molécules à potentiel thérapeutique. Ainsi les connaissances acquises au travers de nos travaux et nos différents modèles nous ont permis de tester différentes molécules telles que des antioxydants comme le N-acétyl-L-Cystéine (NAC) (Segard et al, 2013) ou les tocophérols (Cabet E et al, 2015) mais aussi des molécules pro-autophagique comme le pp242 (Cabet E. et al, 2015) (Figure 6). Nous avons ainsi pu démontrer in cellulo qu’un pré-traitement avec ces molécules diminue l’apparition de l’agrégation des mutants de desmine. Ces résultats sont actuellement en cours d’étude sur le modèle animal (AAV et desKI) mais ouvrent des perspectives thérapeutiques intéressantes.
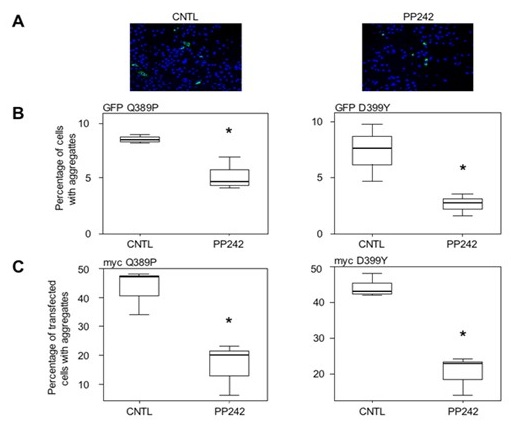
Figure 6 : Effet du traitement au pp242 sur l’agrégation des desmine D399Y et Q389P (Image réalisé avec le microscope inversé Zeiss de la plateforme d’Imagerie BFA)
2- Myopathies Congénitales (Responsable: A. Ferreiro):
A/ SEPN1-related myopathies: Dr Ana FERREIRO, Dr Anne FILIPPE, Dr Maryline MOULIN, Nathalie VADROT
Nous avons une expertise internationale dans le domaine des myopathies à début précoce ( MOE ), qui présentent des troubles musculaires héréditaires hétérogènes avec une faiblesse musculaire dès la naissance ou dans la petite enfance . Nous développons une approche multidisciplinaire pour l’étude des MOE , situé à l’interface avec la recherche clinique.
Dans les dernières années , grâce à une stratégie de collaboration , nous avons défini une ligne de recherche sur les MOE, créé et animé un réseau européen qui a fourni une masse critique de patients, identifiés des mutations dans 4 gènes responsables de 8 formes de MOE, réévalués les phénotypes correspondant et contribué à établir une nouvelle classification nosologique , basé génétiquement dans ce domaine .
Actuellement , nous nous concentrons sur l’obtention d’une meilleure connaissance des mécanismes menant des défauts génétiques à leurs conséquences phénotypiques chez les patients, afin de développer des approches thérapeutiques, notamment pharmacologiques . En particulier, nous avons créé ou contribué à établir que des mutations du gène SEPN1 , codant la sélénoprotéine N (SelN ) , sont responsables de 4 catégories de MOE. En utilisant un modèle ex vivo (cellules en culture de patients ) , nous avons récemment établi que l’oxydation est impliqué dans la physiopathologie de la myopathie SEPN1, et que les antioxydants sont un traitement efficace ex – vivo . Le stress oxydatif induit des modifications épigénétiques dans les cellules souches, et les données préliminaires obtenues par d’autres groupes suggèrent une modulation de marqueurs cellulaires par SelN . Nous sommes intéressés à caractériser davantage ces mécanismes , ainsi que les liens mécaniques entre le stress oxydatif , l’atrophie musculaire sévère observée chez les patients présentant des mutations SEPN1 et la modulation de l’expression de gènes impliqués dans la survie et la mort cellulaire .
Nous allons développer les études pharmacologiques supplémentaires ex – vivo et sur le modèle animal de la carence en SelN , analyser les effets des antioxydants sur les voies mentionnées ci-dessus . Un premier essai clinique de myopathie SEPN1 liés avec des antioxydants est conçu .
B/ ASC-1 related myopathy (TRIP4): Dr Ana FERREIRO, Dr Isabelle DUBAND-GOULET, Dr Eva CABET, Dr Alain LILIENBAUM.
C/ TTN-related myopathies: Dr Ana FERREIRO.

